🧬 Single-Cell Multi-Omics Integration

Stars don't win games. Formations do.
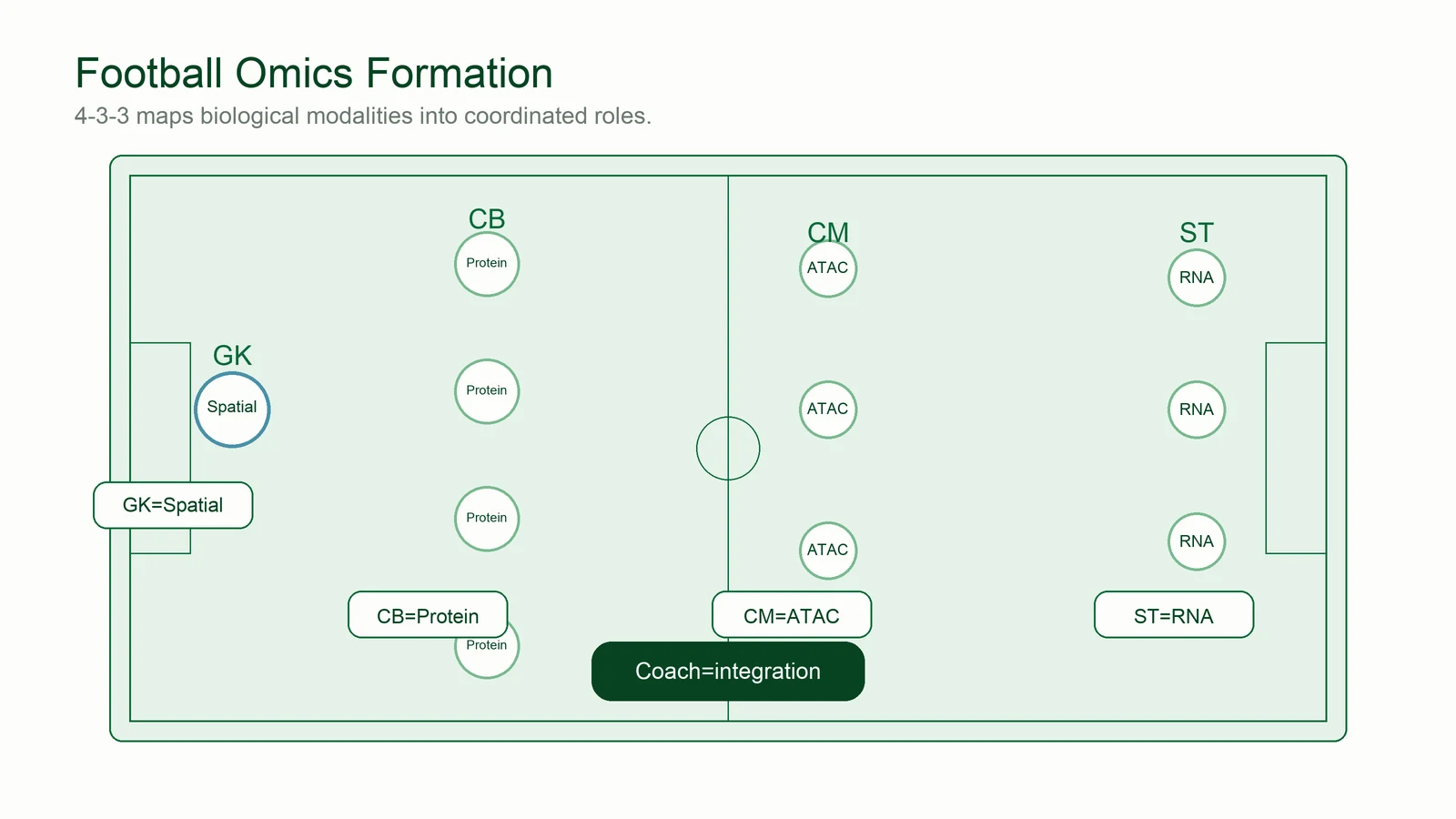
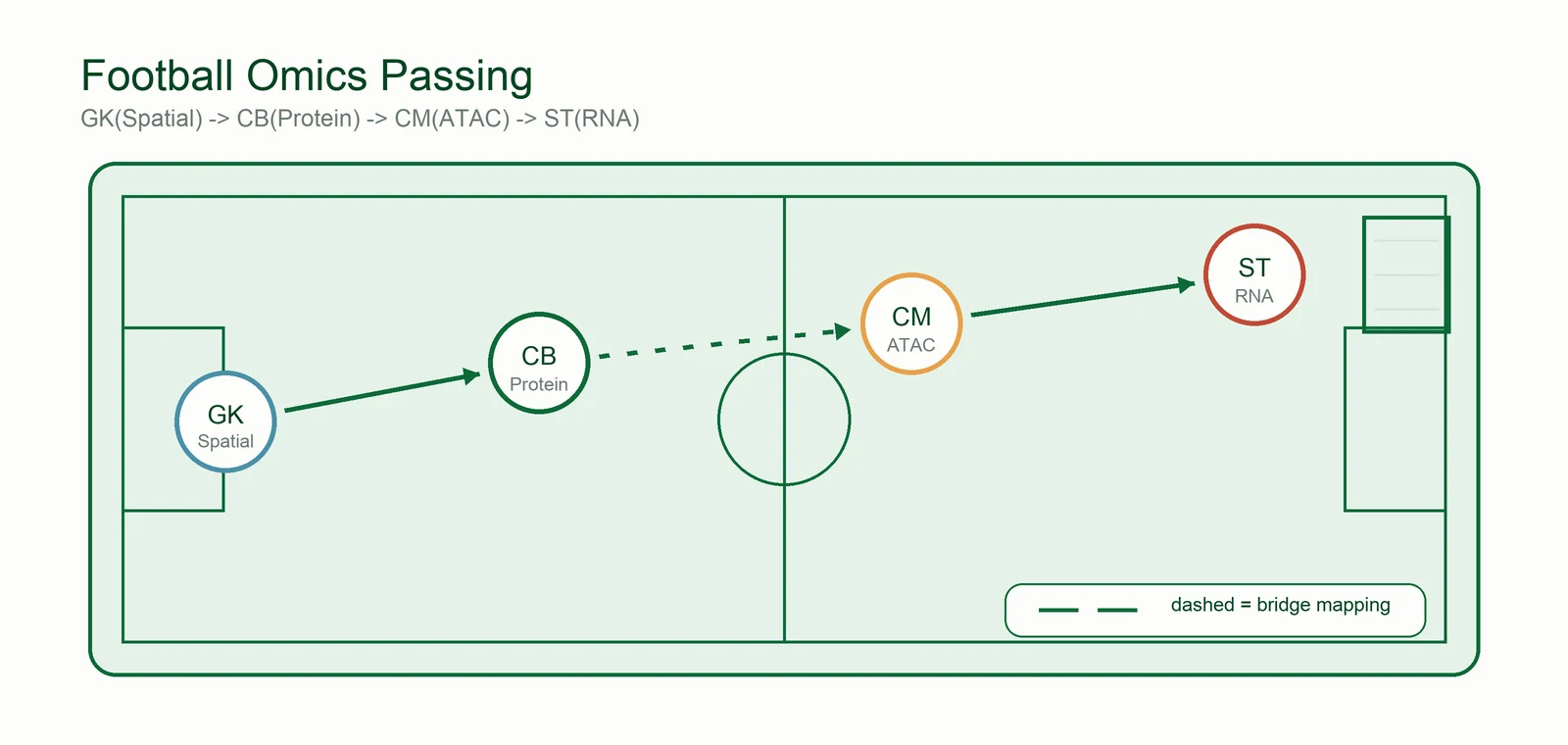
Each omics arrives with its own talent — RNA's speed, ATAC's vision, protein's defense, spatial's positioning. Multi-omics integration is the manager's job: 把十一个天才排成一个阵型 — turn eleven talents into one formation that actually plays together.
Champions Without Stars.
「赢球靠阵型,不靠球星。」
Leicester City, 2015–16: a squad of journeymen, cast-offs and unknowns wins the Premier League at 5000-1 odds — without a single superstar. Six scenes from that season, re-read as multi-omics integration.
The odds nobody believed
Before the season, bookmakers priced a Leicester title at 5000-1 — the same odds as an Elvis sighting. A team with no superstars started winning from day one anyway.
Integration starts here. Not collecting stars, but a formation that already works. See the paradigms ↓
The fastest counter
Eleven straight games with a goal — every time Vardy got the ball it felt like fast-forward, one counter slicing through the whole defensive line.
Transcriptome (scRNA). The fastest, brightest signal on the pitch — but a striker alone never wins a title.
Two lungs, everywhere
The joke went that 71% of Earth is covered by water, and the rest is covered by N'Golo Kanté. He turned up at every duel, stitching defense and attack into one piece.
The midfield is integration itself. The engine that connects every position and lets signals travel. See the methods ↓
The old guard
Two centre-backs in their thirties — no pace, just positioning and cover — cutting off every suspicious pass at the last line.
Protein (CITE-seq). The confirmation layer that settles identity disputes RNA and ATAC argue about.
Position before saves
Schmeichel's numbers were never flashy, but he was already standing in the hardest spot before every shot arrived.
Spatial context. Where a cell sits in the tissue can matter more than how much it expresses. See spatial methods ↓
A different plan for every opponent
Possession one week, counter-attack the next; rotations that changed the shape without breaking it. Ranieri prepared a bespoke answer for every opponent.
Bridge integration. The system adapts — aligning the same team to every new dataset and every new species. See the methods ↓
5000-1 的赔率,0 个球星,1 座冠军 —— integration wins games, not any single modality.
📖 What is Single-Cell Multi-Omics Integration?
Single-cell multi-omics integration combines measurements of different molecular layers (transcriptome, epigenome, proteome, spatial location) from the same or related cells to build comprehensive cellular maps. This integration is essential for understanding cell states, developmental trajectories, disease mechanisms, and therapeutic responses.
Why Multi-Omics Integration Matters
- Comprehensive Cell State Definition: Single modalities provide incomplete views; RNA tells what's transcribed, ATAC reveals accessible chromatin, proteins show functional output
- Regulatory Mechanism Discovery: Linking chromatin accessibility → transcription → protein abundance reveals gene regulatory networks and signaling cascades
- Batch Effect Correction: Harmonizing data across experiments, technologies, and labs enables atlas-scale analyses and meta-studies
- Missing Modality Imputation: Predicting unmeasured features (e.g., protein from RNA) reduces experimental costs while maintaining biological insights
- Spatial Context Integration: Combining molecular profiles with spatial locations reveals tissue architecture and cell-cell interactions
- Perturbation Response Modeling: Understanding how genetic or chemical perturbations affect multiple molecular layers simultaneously
Integration Paradigms (Fu, Shaliu, et al. Nature Methods, 2025), (Liu, Chunlei, et al. Nature Methods, 2025)
According to the benchmark papers, there are six major multi-omics integration paradigms:
🔵 Vertical Integration
🟢 Diagonal Integration
🟡 Mosaic Integration
🔴 Cross Integration
🟣 Spatial Integration
🟠 Perturbation Integration
Each paradigm addresses different data structures and analytical challenges in multi-omics analysis.
📅 Evolution Timeline: From Paired Measurements to Foundation Models
Key Innovations:
- G&T-seq (2015): First simultaneous RNA + DNA methylation
- CITE-seq (2017): RNA + surface protein via antibody tags
- mixOmics (2017): Statistical framework for multi-block data
Era Characteristic: Experimental methods development; simple statistical integration
Key Innovations:
- DIABLO (2019): Multi-omics discriminant analysis
- MOFA+ (2020): Multi-omics factor analysis with covariates
Era Characteristic: Matrix factorization; interpretable latent factors; limited scalability
Key Innovations:
- totalVI (2021): VAE for RNA + protein integration
- Seurat WNN (2021): Weighted nearest neighbor multi-modal analysis
- Concerto (2022): Contrastive learning for 10M+ cells
Era Characteristic: VAE dominance; scalability improvements; atlas-scale analyses
Key Innovations:
- CellOT (2023): Neural optimal transport for perturbations
- SIMBA (2023): Graph embedding with cells + features co-embedded
Era Characteristic: Theoretical rigor; optimal transport theory
Key Innovations:
- scGPT (2024): 100M parameter transformer on 33M cells
Era Characteristic: Pre-training paradigm; 10M+ cell datasets; transfer learning
Key Innovations:
- CellWhisperer (2025): Instruction-tuned multimodal foundation model
- Nicheformer (2025): Spatial multi-omics foundation model
- OmiCLIP (2025): Visual-omics foundation model (H&E + transcriptomics)
- MORPH (2025): Cross-condition perturbation prediction
Era Characteristic: Task-specific foundation models; comprehensive benchmarking; clinical translation focus
🔬 Method Taxonomy: Algorithmic Approaches
By Computational Framework
🧠 Variational Autoencoders (VAE-based)
Principle: Learn probabilistic latent representations with encoder-decoder architecture
Advantages: Uncertainty quantification; generative modeling; missing data imputation
🔄 Contrastive Learning
Principle: Learn representations by pulling similar samples together, pushing dissimilar apart
Advantages: Scalability to millions of cells; no explicit pairing needed; robust embeddings
📊 Graph Neural Networks (GNN)
Principle: Model cells as graph nodes; aggregate information from neighborhoods
Advantages: Captures cell-cell relationships; flexible message passing; spatial awareness
🚀 Optimal Transport
Principle: Find minimal-cost mapping between cell distributions
Advantages: Theoretical guarantees; preserves distributional structure; interpretable
🤖 Foundation Models (Transformers)
Principle: Pre-train large models on massive datasets; fine-tune for specific tasks
Advantages: Transfer learning; few-shot adaptation; generalizable representations
🔗 Matrix Factorization & Classical
Principle: Decompose data matrices into latent factor representations
Advantages: Interpretable factors; computationally efficient; well-understood theory
By Scale Capability
Scalability Tiers
- Small Scale (<10K cells): MOFA+, DIABLO, mixOmics - ideal for pilot studies
- Medium Scale (10K-100K cells): Seurat WNN, totalVI, MultiVI - standard analyses
- Large Scale (100K-1M cells): Concerto, SIMBA, scBridge - atlas construction
- Atlas Scale (>1M cells): Foundation models (scGPT, CellWhisperer), SnapATAC2 - population studies
📄 Landmark Papers by Computational Framework (2015-2025)
🧬 Experimental Technologies (Foundation)
CITE-seq: Simultaneous epitope and transcriptome measurement in single cells
- Antibody-oligonucleotide conjugation method
- Validated on PBMC immune cell populations
- Foundation for multi-modal single-cell biology
scONE-seq: A single-cell multi-omics method enables simultaneous dissection of phenotype and genotype heterogeneity from frozen tumors
- Works with frozen tissue samples
- Simultaneous DNA and RNA profiling
- Tumor heterogeneity analysis
🔷 Variational Autoencoders & Probabilistic Models
Probabilistic harmonization and annotation of single-cell transcriptomics data with deep generative models (totalVI)
- Batch correction across technologies
- Protein imputation from RNA
- Uncertainty quantification
Multi-resolution deconvolution of spatial transcriptomics data reveals continuous patterns of inflammation (MultiVI)
- Handles RNA+ADT+ATAC combinations
- Missing modality imputation
- Spatial deconvolution capabilities
Cobolt: integrative analysis of multimodal single-cell sequencing data
- Hierarchical latent variable model
- Handles incomplete modality measurements
- Supports SNARE-seq and other multimodal data
🕸️ Graph-Based Methods
Integrated analysis of multimodal single-cell data (Seurat WNN)
- Cell-specific modality weighting
- Works with RNA+ADT, RNA+ATAC
- Integrated into widely-used Seurat package
SIMBA: single-cell embedding along with features
- Unified cell-feature embedding space
- Multi-omics native support
- Scales to 1.3M cells in 1.5 hours
🔄 Optimal Transport Methods
Learning single-cell perturbation responses using neural optimal transport (CellOT)
- Drug response prediction
- Genetic knockout effects
- Cross-patient generalization
Cross-modality matching and prediction of perturbation responses with labeled Gromov-Wasserstein optimal transport
- Incorporates perturbation labels
- RNA → protein prediction
- Dose-response preservation
🧠 Deep Learning & Neural Networks
Deep learning and alignment of spatially resolved single-cell transcriptomes with Tangram
- Works with any spatial technology (MERFISH, Visium, smFISH)
- Expands gene coverage to genome-wide scale
- Automated histological registration module
scJoint integrates atlas-scale single-cell RNA-seq and ATAC-seq data with transfer learning
- Processes 1M+ cells in 2 hours
- 84% label transfer accuracy
- Effective batch correction across platforms
scBridge embraces cell heterogeneity in single-cell RNA-seq and ATAC-seq data integration
- Progressive integration strategy
- Exploits cell heterogeneity as advantage
- Superior performance on challenging datasets
scMODAL: a general deep learning framework for comprehensive single-cell multi-omics data alignment with feature links
- GAN-based architecture for alignment
- Works with limited feature correlations
- Cross-modality imputation and prediction
A visual-omics foundation model to bridge histopathology with spatial transcriptomics (OmiCLIP)
- Image → transcriptomics prediction
- Tissue section alignment
- Cell type annotation from H&E
- Spatial decomposition
🎯 Contrastive Learning
Contrastive learning enables rapid mapping to multimodal single-cell atlas of multimillion scale (Concerto)
- 10M cell reference atlas in 1.5 hours
- Query mapping in 8 seconds (10K cells)
- Superior clustering and classification
🤖 Foundation Models & Transformers
scGPT: toward building a foundation model for single-cell multi-omics using generative AI
- Cell type annotation
- Batch correction
- Perturbation prediction
- Gene network inference
CellWhisperer: An instruction-tuned foundation model for single-cell multimodal analysis
- Natural language biological queries
- Multi-task learning (classification, clustering, prediction)
- Zero-shot generalization
🌉 Mosaic & Bridge Integration
Stabilized mosaic single-cell data integration using unshared features (StabMap)
- Multi-hop integration without direct feature overlap
- Leverages non-overlapping features
- Supports supervised and unsupervised modes
Building a cross-species cell atlas with interpretable deep learning (Dictionary Learning)
- Human-mouse integration
- Cross-platform harmonization
- Conserved program discovery
🔬 Perturbation & Response Prediction
Predicting cell morphological responses to perturbations using generative modeling (IMPA)
- Drug response prediction from imaging
- Handles batch effects in HCS
- Generative modeling for perturbation screens
📊 Benchmark & Review Papers
Multitask benchmarking of single-cell multimodal omics integration methods
- No universal winner; task-dependent performance
- Deep learning dominates diagonal/cross integration
- Batch correction often trades off with biological preservation
Benchmarking single-cell multi-modal data integrations
- Multi-gradient AUC for robustness
- Hardware scalability testing (500GB RAM, 24h limits)
- Cross-modality imputation assessment
How to build the virtual cell with artificial intelligence: Priorities and opportunities (AIVC)
- Universal representations (URs) across scales
- Virtual instruments (manipulators & decoders)
- Foundation model architecture for cells
The Human Cell Atlas: from a cell census to a unified foundation model
🔧 Methods Development & Innovation
SnapATAC2: A fast, scalable and versatile tool for analysis of single-cell omics data
- O(n) complexity vs O(n²) for competitors
- 63.4% cost reduction vs ArchR
- 200K cells in 13.4 minutes
SIMVI disentangles intrinsic and spatial-induced cellular states in spatial omics data
- Asymmetric regularization for identifiability
- Single-cell spatial effect estimation
- Causal inference integration (DML)
scMODAL: a general deep learning framework for comprehensive single-cell multi-omics data alignment
- 29-34% imputation improvement
- Works with minimal feature links
- Integrates CITE-seq + CyTOF
MORPH predicts the single-cell outcome of genetic perturbations across conditions and data modalities
- Cross-cell line transfer learning
- Combinatorial perturbation modeling
- Active learning for experiment design
MetaQ: fast, scalable and accurate metacell inference via single-cell quantization
- 100K cells in 0.3h vs 26.7h (SEACell)
- 88% balanced accuracy vs 84% (baseline)
- Native multi-omics support
ADTnorm: robust integration of single-cell protein measurement across CITE-seq datasets
- Cross-institutional integration
- Antibody titration optimization
- Auto-gating (80-100% accuracy)
📚 Comprehensive Method Comparison
By Integration Category & Performance
| Method | Year | Category | Modalities | Scale | Key Strength |
|---|---|---|---|---|---|
| Vertical Integration (Paired Multi-Modal) | |||||
| Seurat WNN | 2021 | Vertical | RNA+ADT, RNA+ATAC | ~100K cells | Cell-specific modality weighting; widely adopted |
| totalVI | 2021 | Vertical/Cross | RNA+ADT | ~50K cells | Probabilistic; batch correction; imputation |
| Multigrate | 2024 | Vertical/Cross | RNA+ADT+ATAC | ~100K cells | Tri-modal support; robust performance |
| Diagonal Integration (Unpaired, Non-Overlapping) | |||||
| scBridge | 2023 | Diagonal | RNA+ATAC | ~50K cells | Superior dimensionality reduction & clustering |
| GLUE | 2022 | Diagonal | RNA+ATAC | ~50K cells | Graph neural network; best batch correction |
| scJoint | 2022 | Diagonal | RNA+ATAC | ~100K cells | Multi-batch integration; transfer learning |
| Mosaic Integration (Overlapping Incomplete) | |||||
| StabMap | 2023 | Mosaic | Any combination | ~50K cells | Flexible; efficient; handles any modality pattern |
| MultiVI | 2023 | Mosaic | RNA+ADT+ATAC | ~100K cells | VAE-based; missing modality imputation |
| Cobolt | 2023 | Mosaic | RNA+ADT+ATAC | ~50K cells | Bayesian framework; uncertainty quantification |
| Spatial Integration | |||||
| SIMVI | 2025 | Spatial | Spatial transcriptomics | ~60K cells | Disentangles intrinsic vs spatial variation |
| OmiCLIP | 2025 | Spatial | H&E + ST | 2.2M pairs | Visual-omics foundation model; H&E → gene expression |
| Tangram | 2021 | Spatial | Spatial mapping | ~50K cells | Maps scRNA-seq to spatial coordinates |
| Perturbation-Aware Integration | |||||
| CellOT | 2023 | Perturbation | RNA-seq (protein/imaging) | ~50K cells | Neural OT; single-cell predictions |
| MORPH | 2025 | Perturbation | RNA + Imaging | ~300K cells | Cross-modality; cross-cell line transfer |
| Labeled GWOT | 2025 | Perturbation | RNA + Protein | ~50K cells | Label-constrained OT; L-fold speedup |
| Classical/Statistical Methods | |||||
| MOFA+ | 2020 | Vertical | Any | ~10K cells | Interpretable factors; handles covariates |
| mixOmics | 2017 | Vertical | Any | ~5K cells | Multiblock projection to latent structure (PLS); statistical rigor |
💡 Practical Implementation Guide
Choosing the Right Method: Decision Framework
Step 1: Identify Your Data Structure
- All cells have all modalities? → Vertical integration
- Different batches, different modalities? → Diagonal
- Mixed modality availability? → Mosaic
- Multiple batches, all modalities? → Cross
- Spatial data? → Spatial methods
- Perturbation data? → Perturbation-aware
Step 2: Consider Your Computational Resources
Limited Resources (no GPU, <32GB RAM):
- Classical methods
Moderate Resources (GPU optional, 32-64GB RAM):
- Most VAE-based and graph methods
High-End Resources (GPU required, 64GB+ RAM):
- Foundation models
Common Pitfalls & Best Practices
- Data Quality First: Ensure proper preprocessing (UMI counts >500, doublet removal, quality control). Use standard pipelines (Scanpy, Seurat). Bad data → bad integration.
- Modality-Specific Normalization: Each modality needs appropriate normalization (RNA: log-normalization, ATAC: TF-IDF, Protein: ADTnorm/DSB). Don't use RNA normalization for ATAC!
- Evaluation Metrics Matter: Use multiple complementary metrics (ARI, NMI, ASW for clustering; kBET, iLISI for batch mixing). Single metrics can be misleading.
- Batch Effect vs Biological Variation: Over-correction removes biology; under-correction leaves technical artifacts. Check cell type separation alongside batch mixing.
- Held-Out Testing: Evaluate on held-out batches or cell types, not just held-out cells from training conditions. Tests true generalization.
- Baseline Comparisons: Always compare against simple baselines (PCA, naive concatenation, per-modality analysis). Complex methods should outperform simple ones.
- Computational Scalability: Test methods on your actual data size. Many methods fail silently or degrade performance on large datasets.
- Biological Validation: Computational integration should reveal biologically meaningful patterns. Validate key findings with marker genes, known cell types, or experimental follow-up.
- Hyperparameter Tuning: Default parameters often suboptimal. Use cross-validation or grid search for critical parameters (latent dimensions, regularization, learning rate).
- Reproducibility: Report random seeds, package versions, preprocessing steps, and hyperparameters. Provide code and (when possible) data.
Software Ecosystem & Tools
- Scanpy: Standard Python toolkit for preprocessing, analysis, visualization. Foundation for many integration methods.
- Seurat: R package with Seurat v3/v5 integration, WNN, UMAP. Industry standard for single-cell RNA-seq.
- scvi-tools: PyTorch-based framework for probabilistic models (scVI, totalVI, MultiVI, scANVI). GPU-accelerated.
- Muon: Python package for multimodal omics analysis. Integrates with Scanpy ecosystem.
- LIGER: R package for integrative non-negative matrix factorization. Good for cross-platform integration.
- Harmony: Fast batch correction algorithm. Works on PCA embeddings. Widely used for large-scale integration.
⚖️ Side-by-Side: Combining Modalities — Weighted-Graph (WNN) vs Joint Generative (totalVI / MultiVI)
When RNA and a second modality (protein in CITE-seq, chromatin in 10x Multiome) are measured in the same cell, the integration question is when to combine them: fuse per-modality neighbor graphs after embedding each separately (WNN), or learn a single joint latent space from the raw counts of both (a joint VAE like totalVI / MultiVI)?
The same paired cells become one integrated representation in two different ways, depending on whether modalities are fused late (graph) or early (latent):
The root decision is fuse graphs after separate embeddings vs learn one joint latent from raw counts. WNN is model-free and hands you an interpretable per-cell weight showing which modality drives each cell, but it does no denoising or imputation; totalVI/MultiVI is probabilistic — it denoises, imputes missing modalities, and corrects batch — at the cost of training a model. Both assume paired cells; for unpaired / diagonal data (different cells per modality) you need anchor transfer, GLUE, or LIGER instead.
🛠️ Hands-On Practice
The steps below walk through the joint-VAE route on CITE-seq with totalVI — one shared latent from RNA + protein — and note the WNN (graph) alternative and the MultiVI variant for RNA + ATAC.
Environment & packages
scvi-tools provides totalVI/MultiVI; scanpy/muon/mudata hold the modalities and run the WNN graph route in Python (Seurat's WNN is the R equivalent).
conda create -n multiomics python=3.10 -y
conda activate multiomics
pip install scvi-tools scanpy muon mudata
# WNN graph route is in muon (Python) or Seurat (R)Hardware. WNN runs on a laptop; totalVI/MultiVI training wants a GPU (minutes) but works on CPU (slower) for datasets up to ~100k cells.
Data structures & formats
- CITE-seq
AnnData— RNA counts inadata.X, protein (ADT) counts inadata.obsm["protein_counts"] - 10x Multiome
MuData—mdata["rna"](genes) +mdata["atac"](peaks), the same cells in both - Paired requirement — totalVI, MultiVI, and WNN all need the same cells measured across modalities
- Joint latent — written to
adata.obsm["X_totalVI"]/["X_multivi"]for neighbors + UMAP - Batch key —
adata.obs["batch"](donor / lane) for the model to correct
Minimal code walkthrough
Fit totalVI on CITE-seq to get one joint latent from RNA + protein.
import scvi, scanpy as sc
# CITE-seq: RNA counts in adata.X, protein (ADT) counts in adata.obsm["protein_counts"]
adata = sc.read_h5ad("cite_seq.h5ad")
# totalVI jointly models RNA (negative binomial) and protein (with ambient background)
scvi.model.TOTALVI.setup_anndata(
adata,
protein_expression_obsm_key="protein_counts",
batch_key="batch",
)
model = scvi.model.TOTALVI(adata)
model.train()
# One joint latent from BOTH modalities; denoised protein is available too
adata.obsm["X_totalVI"] = model.get_latent_representation()
sc.pp.neighbors(adata, use_rep="X_totalVI"); sc.tl.umap(adata)Swap in the graph route, or MultiVI for RNA + ATAC:
# Graph-level alternative — WNN via muon (Python port of Seurat's WNN):
# import muon as mu
# mu.pp.neighbors(mdata) # per-modality graphs -> one weighted joint graph
# mu.tl.umap(mdata)
# RNA + ATAC (10x Multiome) instead of CITE-seq -> MultiVI:
# amvi = scvi.data.organize_multiome_anndatas(adata_paired, adata_rna, adata_atac)
# scvi.model.MULTIVI.setup_anndata(amvi, batch_key="modality")
# mvi = scvi.model.MULTIVI(amvi, n_genes=n_g, n_regions=n_r); mvi.train()Common pitfalls & tips
- Paired vs unpaired. totalVI / MultiVI / WNN require the same cells in every modality; for different cells per modality (diagonal integration) use Seurat anchor transfer, GLUE, or LIGER.
- Never concatenate raw features and PCA. RNA counts, protein ADT, and sparse binary ATAC peaks live on wildly different scales — naive concatenation lets one modality dominate the embedding.
- Protein has ambient background. ADT counts include background binding; totalVI models it explicitly, so don't preprocess protein like RNA (use CLR for WNN, raw counts for totalVI).
- ATAC needs its own preprocessing. TF-IDF / binarization and peak calling come first; feeding raw peak counts into an RNA-style model is wrong.
- Correct batch and modality. Donor / lane batch effects exist within each modality — set the batch key, not just the modality label.
- Inspect the WNN weights. The per-cell modality weights are interpretable; a modality with no signal in some cells should get a low weight — check them to catch a broken or empty modality.